


Mutatie specifieke behandelingen
Exon skipping en stop codon readthrough
Exon skipping en stop codon readthrough zijn mutatie specifieke technieken. Dit betekent dat ze enkel voor een subset patiënten met een specifieke mutatie zullen werken (zie onderstaand voor meer informatie). Om te weten of een Duchenne patiënt in aanmerking komt voor exon skipping of stop codon readthrough is het belangrijk om exact te weten wat voor soort mutatie de ziekte heeft veroorzaakt.
Exon skipping
Voor Duchenne spierdystrofie
Stand van zaken
Vier exon skipping medicaties zijn momenteel goedgekeurd in de VS (eteplirsen, golodirsen, casimersen en viltolarsen) en een in Japan (viltolarsen). Deze middelen hebben versnelde goedkeuring ontvangen gebaseerd op hun vermogen tot het herstellen van lage levesl dystrofine. Studies zijn nog gaande om aan te tonen of deze levels aan dystrofine ook de ziekteprogressie vertragen.
Doel
De genetische code te corrigeren en de productie van een gedeeltelijk functioneel dystrofine mogelijk te maken.
Achtergrond
De genetische code van genen is verspreid over zogenaamde exonen. Wanneer een eiwit gemaakt moet worden, maken genen een tijdelijke kopie (RNA genaamd). Voordat dit RNA in eiwit kan worden vertaald, moeten de exonen eerst gekoppeld worden en de tussenstukken die de genetische code (introns) niet bevatten, moeten verwijderd worden. Dit is een proces dat “splicing” genoemd wordt.
Bij Duchenne-patiënten wordt de genetische code van het DMD-gen verstoord, wat betekent dat de code onleesbaar wordt. Dit resulteert erin dat de vertaling van gen naar eiwit voortijdig afgebroken wordt. In Becker-patiënten behouden mutaties de genetische code, wat een de productie van een eiwit dat de functionele domeinen behoudt, mogelijk maakt.
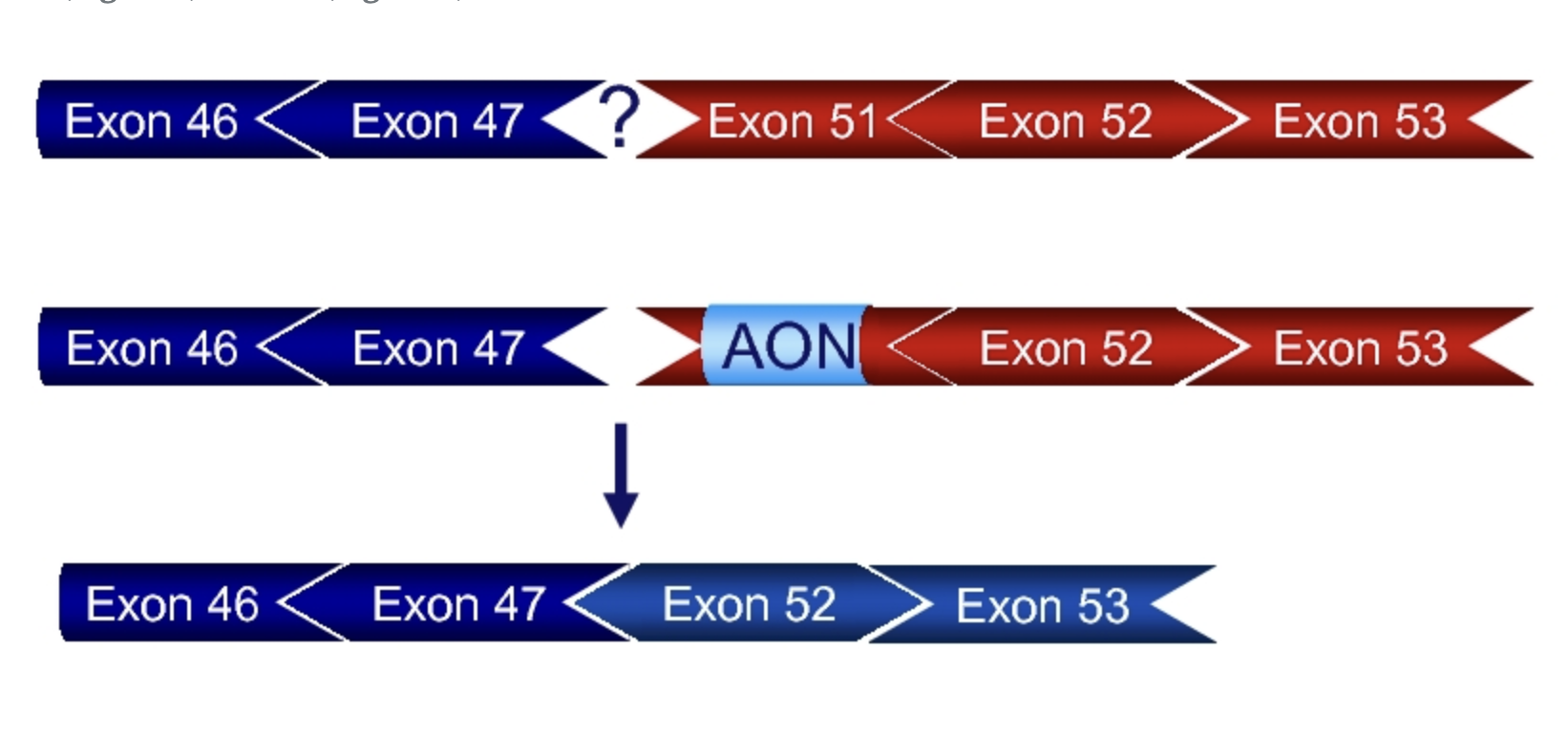
Exon skipping heeft tot doel de genetische code van Duchenne-patiënten te herstellen, zodat er een gedeeltelijk functioneel, Becker-achtig dystrofine-eiwit kan worden gemaakt in plaats van een niet-functioneel Duchenne-eiwit. Dit wordt bereikt met behulp van AONs (antisense oligonucleotiden). AONs zijn kleine stukjes gemodificeerd RNA die een specifiek exon herkennen, daaraan binden en het voor de splicingmachines verbergen. Dit resulteert in het skippen van dat exon en het herstel van de genetische code.
Annemieke Aartsma-Rus legt exon skipping uit in dit filmpje.
Exon skipping wordt ook uitgelegd in deze video ‘Dance your PhD’.
AON-behandeling heeft geleid tot exon skipping resulterend in de productie van Beckerachtige dystrofines in gekweekte cellen afkomstig van patiënten en het mdx-muismodel. In mdx-muizen ging dit gepaard met functionele verbetering.
AON-middelen maken
AONs zijn kleine stukjes gemodificeerd DNA. De modificaties zijn nodig om de AONs geneesmiddelachtige eigenschappen te geven, bijv. om ervoor te zorgen dat ze resistent zijn tegen enzymen die DNA afbreken, waardoor ze door weefsels kunnen worden opgenomen en om te voorkomen dat ze door de nieren worden uitgefilterd.
Toepasbaarheid
Voor verschillende mutaties en soorten mutaties moeten verschillende exonen worden geskipt om de genetische code te herstellen. Omdat de meeste patiënten een deletie hebben en deze zich in een hotspot bevinden, is het skippen van sommige exonen van toepassing op meer patiënten dan anderen. Een schematische presentatie van de exonen in het DMD-gen is hier beschikbaar. Tot slot, helpt de DOVE-tool bij het bepalen welk exon geskipt moet worden voor een bepaalde mutatie.
Hoewel exon-skipping gunstig zou zijn voor de meeste mutaties, zijn er enkele uitzonderingen.
Samenvatting van de huidige situatie
Goedgekeurde AONs
Er zijn momenteel 4 AON’s goedgekeurd. Deze zijn van de phosphorodiamidate morpholino oligomer (PMO) chemie. Dit is een chemie die zeer efficiënt door de nieren wordt uitgefilterd. Daarom is frequente behandeling met hoge doses AON’s nodig (wekelijkse intraveneuze injecties van 30-80 mg/kg) en de opname door de skeletspieren is suboptimaal, wat resulteert in slechts een minimale toename van dystrofine. Om de levering aan de skeletspieren en het hart te verbeteren, worden verschillende benaderingen bestudeerd:
- Testen van chemische aanpassingen in de medicatie die voorkomen dat de nieren de medicatie wegfilteren en die betere werkzaamheid en veiligheid vertonen dan vergelijkbare verbindingen die eerder zijn ontwikkeld (zie drisapersen).
- Een positief geladen peptide aan PMO’s koppelen. Dit zal de levering aan alle weefsels verbeteren, inclusief aan spieren en hart. Het nadeel is dat de positief geladen peptiden tot toxiciteit kunnen leiden. De vraag is: wat komt eerst: Verbeterde efficiëntie of toxiciteit.
- Het koppelen van een conjugaat aan PMO’s die de specifieke opname door skeletspieren verbetert. Dit kan worden bereikt met antilichamen voor de transferrinereceptor, die in verrijkte aantallen in de skeletspieren voorkomt.
Hieronder bekijk ik de ontwikkeling per exon.
Ter overweging
Aangezien exon skipping erop gericht is om de aanmaak van dystrofine te herstellen, gebruiken geneesmiddelenontwikkelaars de analyse van dystrofine na behandeling om te beoordelen of de therapie het beoogde effect heeft gehad. Omdat de meeste Duchenne-patiënten lage, maar individueel variabele hoeveelheden dystrofine produceren, moet de vergelijking worden gemaakt tussen het dystrofineniveau na behandeling en het uitgangsniveau (baseline) om vast te stellen of de hoeveelheid is toegenomen.
Dit wordt meestal gedaan met een techniek die Western blotting wordt genoemd. Hierbij wordt de hoeveelheid en de lengte van dystrofine gemeten ten opzichte van een referentiemonster (menselijke controlespier). Gezien de hoeveelheid dystrofine in controlespieren kan variëren, is het moeilijk om dystrofineniveaus tussen verschillende ontwikkelaars te vergelijken. Daarnaast beïnvloedt het tijdstip waarop het biopt wordt genomen hoeveel dystrofine wordt aangemaakt, wat per onderzoek kan verschillen.
Tot slot zijn sommige exons makkelijker over te slaan dan andere. Hierdoor is het lastig om dystrofineniveaus te vergelijken tussen AON’s die verschillende exons targeten en tussen klinische studies van verschillende bedrijven.
Bevestigende onderzoeken voor de goedgekeurde exon-skippingmiddelen (PMO’s)
Phosphorodiamidate Morpholino Oligomeren
Eteplirsen (Exon 51 skipping)
Één exon skipping AON van de morfolino (PMO)-chemie, eteplirsen (exondys51) genaamd, heeft versnelde goedkeuring van de FDA gekregen. Dit was uitsluitend gebaseerd op herstel van dystrofine. Het bedrijf dat eteplirsen (Sarepta) ontwikkelt, moet nog steeds bevestigen dat de behandeling de ziekteprogressie vertraagt. Eteplirsen is niet goedgekeurd door de EMA. In juni 2018 gaf de Committee for Human Medical Products (CHMP) van de EMA een negatieve beoordeling, waarna Sarepta in beroep ging. Dit beroep is echter afgewezen in september 2018.
Klinische studies met Eteplirsen
Eteplirsen is een PMO AON die gericht is op exon 51. Het moet intraveneus worden toegediend. Eteplirsen werd getest bij 19 patiënten in verschillende doses tot 20 mg/kg. Omdat niet alle patiënten in deze studie even goed reageerden, werd kleine vervolgstudie uitgevoerd met 12 patiënten waarbij twee hogere doses werden getest. In deze studie werd de dystrofine productie hersteld voor alle patiënten na 24 weken behandeling met eteplirsen. De patiënten zijn inmiddels meer dan 188 weken behandeld en voor de tien patiënten die nog steeds ambulant zijn, daalde de zes minuten loopafstand minder dan verwacht zou worden uit het natuurlijke ziektebeloop (hoewel dit met de nodige voorzichtigheid moet worden geïnterpreteerd vanwege de kleine groepsgrootte).
De FDA kondigde op 19 september 2016 aan dat eteplirsen versnelde goedkeuring kreeg. Dit was alleen gebaseerd op kleine stijgingen in dystrofineniveaus waargenomen in spierbiopten van behandelde patiënten. De FDA heeft aangegeven dat functionele effecten nog niet zijn bevestigd. Als zodanig moet Sarepta het klinische voordeel bevestigen in aanvullende klinische studies die momenteel aan de gang.
Een fase 3 studie waarbij wekelijkse intraveneuze toediening met 30 mg/kg eteplirsen gedurende 96 weken in ambulante patiënten wordt getest, is afgerond in de VS en de resultaten zijn gepubliceerd. Dit was een open-labelstudie waarin patiënten met mutaties, waarbij exon 51 skipping mogelijk is, behandeld werden, terwijl patiënten met niet-behandelbare mutaties werden gebruikt als controles voor functionele testen en veiligheid. Het was helaas niet mogelijk om de onderzoeksgroepen te vergelijken omdat de meeste patiënten in de controlegroep uit het onderzoek stapten voordat het afgerond was.
Een klinische studie in Europa in patienten tussen de 6 maanden en 4 jaar oud is voltooid en laat zien dat de behandeling goed getolereerd wordt in jonge patienten.
Bovendien zijn open-labelstudies bij jonge patiënten (jonger dan zes jaar) en bij beperkt- of niet-ambulante patiënten in de VS gestart. In het onderzoek bij jonge patiënten wordt opnieuw een groep met niet-ontvankelijke mutaties als controle gebruikt. Tot slot loopt er ook een nieuwe klinische studie waarin hogere doseringen eteplirsen getest worden (100 en 200 mg/kg/week), zoals verzocht door de FDA.
Golodirsen (exon 53 skipping)
Sarepta heeft een studie afgerond met PMOs gericht tegen exon 53 (golodirsen, samenwerking met Francesco Muntoni in Londen). Na 48 weken behandeling werd een toename in dystrofine-expressie van ~1% waargenomen. Op basis hiervan heeft Sarepta in het vierde kwartaal van 2018 goedkeuring bij de FDA aangevraagd voor golodirsen, In augustus 2019 heeft de FDA aan Sarepta laten weten dat golodirsen om veiligheidsredenen niet goedgekeurd werd, namelijk dat in preklinische dierstudies nierschade gezien werd na toediening van hoge doses golodirsen en de infectierisico’s van intraveneuze katheders die nodig zijn voor de herhaalde intraveneuze infusies. Nadat Sarepta de aanvraag opnieuw had ingediend en ook de punten waarover men zorgen had aangepakt had, keurde de FDA golodirsen in december 2019 goed. Net als bij eteplirsen benadrukte de FDA dat de functionele effecten van de behandeling met golodirsen nog moeten worden bevestigd. Momenteel loopt er een klinische studie om de functionele effecten van golodirsen te bepalen.
Viltolarsen (exon 53 skipping)
Nippon Shinyaku (Japan) en NS-Pharma hebben klinische studies uitgevoerd met PMOs voor het skippen van exon 53 (viltolarsen) in Japan en in ambulante patiënten in de VS. Na 24 weken behandeling met hoge doseringen (40 en 80 mg/kg) werd in spierbiopten tot 5% dystrofine gemeten. NS-Pharma kreeg goedkeuring van de FDA voor viltolarsen in augustus 2020 en van het Japanse ministerie van Volksgezondheid, Arbeid en Welzijn in maart 2021. Een bevestigende studie werd uitgevoerd waar patienten wekelijkse injecties met viltolarsen (80 mg/kg) of placebo kregen. Het primaire eindpunt, de tijd om op te staan vanaf de vloer, van de studie werd niet behaald, gezien er geen verschillen zichtbaar waren tussen de behandelde patienten en de placebo groep. Deze studie is momenteel bezig met het verzamelen van extra data voor een 2 jaar behandel periode.
Ook is een klinische studie gaande waar ambulante en niet-amublante patienten worden behandeld met viltolarsen.
Casimersen (exon 45 skipping)
Sarepta is een placebogecontroleerde fase 3 studie van 96 weken gestart om exon 45 (casimersen) en 53 AONs te evalueren. Tussentijdse analyse van een spierbiopt van de met casimersen behandelde patiënten liet een toename van dystrofineniveaus zien van 0.9% (baseline) tot 1.7% (na één jaar behandeling). Op basis van deze resultaten keurde de FDA casimersen in februari 2021 goed. Ook hier benadrukte de FDA dat de functionele effecten van casimersen nog moeten worden bevestigd.
Overige studies met PMOs
Brogidirsen (exon 44 skipping)
NS-Pharma is een klinische studie gestart met NS-089 in Japan. Dit is een PMO gericht op exon 44. Een open-label studie werd uitgevoerd in 6 ambulante patienten in Japan. Deze patiënten kregen wekelijks een intraveneuze infusie van 40 mg/kg of 80 mg/kg brogidirsen. Resultaten van de studie zijn gepubliceerd. Patiënten lieten een verhoging van 10% zien in dystrofine levels bij de 40 mg/kg dosis, en een verhoging van 15% voor de 80 mg/kg dosis.
Opvallend is dat patienten die exon 44 skipping nodigh hebben al een hoger basis level aan dystrofine hebben, gezien exon 44 skipping ook spontaan plaatsvindt. Mogelijk zorgt dit proces ervoor dat epxressie van dystrofine na exon 44 skipping verder wordt verhoogd (zie ook AXC1044).
NS Pharma is ook een klinische studie gestart met een PMO voor exon 50 skipping (NS-50/NCNP-03). Er zijn nog geen resultaten bekend.
Exon skipping van een enkele exon duplicatie
Sarepta voert een klinische studie uit voor exon 45, 51 en 53 skipping in patienten met een duplicatie van exon 45 (casimersen), exon 51 (eteplirsen) en exon 53 (golodirsen). Resultaten zijn gepresenteerd op congressen en laten dystrofien restoratie zien na behandeling.
Verbetering van exon skipping met chemische modificaties
BMN351 (exon 51 skipping)
BioMarin ontwikkelt BMN351, een ASO met een andere ruggengraat dan de goedgekeurde exon-skipping geneesmiddelen (fosforothioaat). Dit zorgt voor een betere biologische beschikbaarheid van de AON, waardoor lagere doses kunnen worden gebruikt. Echter, deze ruggengraat kan ook bijwerkingen veroorzaken (zie het drisapersen kopje hieronder). BMN351 leidde tot dystrofineherstel in een muismodel met een gemuteerde versie van het menselijke dystrofinegen. BioMarin voert nu een klinische studie uit met BMN351 bij Duchenne-patiënten.
SQY51 (exon 51 skipping)
Synthena heeft een AON in ontwikkeling met een ‘tricyclo DNA’ chemie die exon 51 target. Er wordt momenteel een klinische studie gehouden in Frankrijkg voor SQY51 in Duchenne patienten.
WVE-N531
Wave voert een klische studie uit met WVE-N531, een ASO met een betere biobeschikbaarheid dan de PMO chemie. Een interne analyse liet zient dat na 24 weken of tweeweekelijkste behandeling met 10 mg/kg WVE-N531, patienten 5.5% meer dystrofine produceerde. De behandeling werd goed getolereerd en patiënten zijn momenteel geincludeerd in een open label studie.
Renadrisen (exon 51 skipping)
Daiichi Sankyo was in Japan AONs met de ENA-chemie tegen exon 45 aan het ontwikkelen. Een eerste studie liet zien dat de AON veilig is, maar dat de toenames in dystrofine waren uiterst laag waren. Daiichi Sankyo heeft aangegeven te stoppen met de ontwikkeling van dit middel.
Verhogen van de weefsel opname met korte peptiden
Vesleteplirsen
Eteplisen (exon 51 skipping) zorg enkel voor een kleine verhoging van de hoeveelheid dystrofine. Er is ruimte voor verbetering van AONs. Een strategie die gebruikt wordt om AONs te verbeteren is door een korte, positief geladen peptide aan de AON te linken. Deze peptide verhoogt de opname van de AON in weefsel, en dus ook in de spieren. Sarepta heeft een variant van eteplirsen getest met een peptide-aanpassing (SRP-5051 of vesleteplirsen). Resultaten van biopten na 12 weeken behandeling via maandelijkse dosering van 30 mg/kg vesleteplisren lieten ongeveer 2% dysfrofine zien in 4 Duchenne patienten. Een verlaging van magnesium in het bloed werd geconstateerd (hypomagnesiëmie) en behandeld met magnesium supplementen. In deel B van de klinische studie werd een grotere groep patienten behandeld met 30 mg/kg vesleteplirsen per maand. Door een nieuw geval van hypomagnesiëmie werd de studie tijdelijk gepauzeerd. Na 7 maanden behandelijk lieten biopten ongeveer 5% dystrofine zien in 20 patienten. Lage concentraties van magnesium en calcium werden gemeten in het bloed en behandeld met supplementen.
In oktober 2024 heeft Sarepta aangekondigd dat ze de ontwikkelingen van vesleteplirsen stoppen in verband met veiligheidsoverwegingen, gezien nierschade tussen de behandelmomenten door niet herstelde en de nierfunctie achteruitging.
PGN-EDO51
PepGen ontwikkeld een vergelijkbare medicatie (peptide gelinkte PMO) genaamd PGN-EDO51. De stof is getest in gezonde vrijwilligers en klinische trials voor Duchenne patienten worden momenteel uitgevoerd. Exon skipping vond plaats in gezonde vrijwilligers en niet-menselijke primaten, echter werd ook hypomagnesiëmie geconstateerd.
Voorlopige resultaten van een dosisbepalingsstudie laten exon 51 skipping zien en een kleine verhoging van dystrofine expressie (0.3%) voor patiënten behandeld met een maandelijkse dosis van 5 mg/kg voor 3 maanden. Doseringen van 10 mg/kg zorgde voor een vergelijkbare verhoging van dystrofine (0.4%), waarna het bedrijf heeft besloten in mei 2025 om te stoppen met de ontwikkeling van PGN-EDO51.
ENTR-601-44
Entrada ontwikkeld ENTR-601-44, een PMO met een EEV peptide gericht op exon 44 skipping. De aminozuren van deze peptide zijn kleiner dan van vesleteplirsen en PGN-EDO51, maar heeft een lagere positieve lading en de peptide is circulair, wat naar verwachting zou moeten zorgen voor lagere toxiciteit. ENTR-601-44 is getest in gezonde vrijwilligers in de UK, waar exon 44 skipping zichtbaar was in een spierbiopt. Tolerantie was goed. Momenteel loopt er een klinische studie in Duchenne patiënten.
ENTR-601-45
Entrada heeft ook ENTR-601-45 ontwikkeld, deze PMO richt zich op exon 45 en heeft dezelfde EEV peptide als ENTR-601-44. Een klinische studie is gepland maar inlcusie is nog niet gestart.
Verhogen van de weefsel opname met transferrine receptor antilichamen
Specifiek aan spieren en hart leveren
AONs kunnen ook specifieker aan de spieren en het hart geleverd worden door de AONs te conjugeren met transferrin receptor antilichamen. De transferrin receptor (TfR1) is een ijzer transporter die zich in bijna alle cellen van het lichaam bevind, maar vooral hoog tot expressie komt in de spiercellen. De antilichamen binden aan de receptor en faciliteren zo de opname van de AON, zonder de verder functie van de receptor te verstoren.
DYNE-251 (exon 51 skipping)
Dyne Therapeutics heeft een PMO met een antilichaam fragment dat herkent wordt door de transferrin receptor in ontwikkeling die exon 51 target, DYNE-251. Resultaten laten zien dat patienten die 6 maanden behandeld werden met een maandelijkse dosis van 5 mg/kg DYNE-251 0.9% meer dystrofine produceerde, terwijl de patienten dei behandeld werden met 10 of 20 mg/kg 3 tot 3.7% meer dystrofine produceerde. Patienten die 40 mg/kg ontvingen ondervonden bijwerkingen, waarschijnlijk gerelateerd aan de DYNE-251. Patienten zijn geheel hersteld van de bijwerkingen. Patienten die voor langere tijd met 10 of 20 mg/kg werden behandeld lieten verbeteringen zien in de spierfunctie, zichtbaar in meerdere uitkomstmaten.
Delpacibart zotadirsen (exon 44 skipping)
Avidity voert een klinische studie uit met delpacibart zotadirsen AXC-1044, een PMO gericht op exon 44, die gelinkt is aan een antilichaam gericht op transferring (voor verbeterde levering aan de spieren, vergelijkbaar met de Dyne strategie, maar met een groter antilichaam). Eerste resultaten laten een 25% verhoging in dystrophine zien na drie 6-wekelijkse intraveneuze behandeling met 5 mg/kg delpacibart zotadirsen. Patiënten met een dosis van 10 mg/kg elke 8 weken lieten geen hoger dystrofine herstel zien. De huidige patiënten in de klinische studie wisselen nu naar een regime van 5 mg/kg elke 6 weken.
Exon skipping genen
Exon skipping ASOs moeten regelmatig (wekelijks of maandelijks) worden toegediend en de
leveringsefficiënte is momenteel relatief laag. Onderzoekers hebben de mogelijkheid onderzocht om met behulp van AAV een antisense-gen aan de spieren te leveren. Hierdoor worden gentherapie en exon skipping gecombineerd.
Kevin Flanigan (Columbus Ohio) heeft een antisense-gen ontwikkeld voor patiënten met een duplicatie van exon 2. Tot nu toe zijn er 3 patiënten van 7 maanden, ~9 jaar en ~14 jaar, behandeld. Biopsieën lieten herstel van dystrofine zien in niveaus van respectievelijk 70%, 6% en 1% na 12 maanden behandeling.
Audentes/Astellas waren klinische onderzoeken met antisense-genen voor het overslaan van exon 51, 53 en 45 aan het voorbereiden. De ontwikkeling kreeg echter onlangs geen prioriteit meer en werd beëindigd.
Andere exonen
Er lopen preklinische onderzoeken bij verschillende bedrijven die actief zijn op het exon skipping gebied om exon skipping middelen voor 52, 54 en 55 te identificeren.
Mutatie specificiteit
AONs die verschillende exonen overslaan, worden door de regelgevende instanties als verschillende geneesmiddelen beschouwd. Dit betekent dat het ontwikkelen van AON’s voor verschillende exonen zeer kostbaar en tijdrovend is, omdat elk exon alle fasen van de preklinische en klinische ontwikkeling moet doorlopen.
Hopelijk zal de ontwikkeling van AON sneller worden na de eerste twee of drie. TREAT-NMD coördineert een dialoog hierover met regelgevende instanties namens exon skipping wetenschappers, artsen en de industrie en de patiënten community. De meest recente bijeenkomst vond plaats op 29 april 2015. De publicatie van de bijeenkomst is beschikbaar (een gratis exemplaar kan hier gevonden worden).
Klinische studies met drisapersen (stopgezet)
Wave Life Sciences heeft een dosisoplopende fase 1 veiligheidsstudie uitgevoerd met de exon 51 skipping AON suvodirsen, die een nieuwe modificatie heeft. Hieruit bleek dat suvodirsen goed verdragen werd bij lagere doses, maar dat de intensiteit van de bijwerkingen (koorts, misselijkheid en hoofdpijn) ernstiger was bij hogere doses. Uit een placebogecontroleerd fase 2/3 onderzoek waarin behandeling op langere termijn met lagere doses suvodirsen werd geëvalueerd, bleek dat suvodirsen niet resulteerde in een verhoging van de dystrofineniveaus in biopsieën. Daarom is de ontwikkeling van suvodirsen stopgezet.
De 2OMePS AON gericht op exon 51 is drisapersen of kyndrisa genoemd. Alle patiënten de betrokken waren bij de vroege subcutane (onderhuidse) studie werden opgenomen in een open-label extensiestudie waar ze wekelijks behandeld werden met drisapersen. Patiënten werden langer dan zes jaar behandeld (inclusief pauzes in de behandeling). Voor de patiënten die bij het begin van de extensiestudie nog ambulant waren, is de zes minuten loopafstand gestabiliseerd, terwijl het natuurlijke ziektebeloop een afname zou voorspelen. Bij gebrek aan een placebogroep moeten deze resultaten echter met voorzichtigheid worden geïnterpreteerd.
GlaxoSmithKline (GSK) had de licentie voor drisapersen van Prosensa overgenomen en heeft verschillende onderzoeken gecoördineerd. Bij alle onderzoeken waarbij onderhuidse injectie van drisapersen werd gebruikt, werden reacties op de injectieplaats en proteïnurie vaker waargenomen bij met drisapersen behandelde patiënten dan bij met placebo behandelde patiënten. Er is studie gedaan waarbij verschillende doseringsschema’s werden vergeleken, in patiënten die zich in een relatief vroeg stadium van de ziekte bevonden. Deze studie omvatte 54 patiënten die gedurende 48 weken placebo, wekelijkse onderhuidse behandeling met drisapersen of een afwisselende behandeling kregen. Beide behandelde groepen liepen ~35 meter meer dan met een placebo behandelde patiënten in de 6 minuten looptest. Een studie waarbij verschillende doses werden vergeleken, is gedaan in patiënten die zich in een vroeg ziektestadium bevonden (in 15 seconden van de vloer konden opstaan). Patiënten kregen gedurende 24 weken placebo, 3 of 6 mg/kg drisapersen. Patiënten behandeld met 6 mg/kg liepen na 24 weken 27 meter meer dan patiënten behandeld met placebo of 3 mg/kg.
In 2011 werd een fase 3 placebogecontroleerde studie gestart om de veiligheid en effectiviteit van behandeling met drisapersen bij 186 ambulante patiënten te testen. Er werd geen significant verschil in de 6 minuten looptest waargenomen tussen placebo en patiënten behandeld met drisapersen na 48 weken. Ondertussen heeft GSK de licentie om drisapersen te ontwikkelen teruggegeven aan Prosensa en Prosensa is overgenomen door BioMarin.
Prosensa/BioMarin hebben de verzamelde gegevens van de systemische studies en extensiestudies geanalyseerd. De resultaten wijzen op een tragere ziekteprogressie bij behandelde jongere patiënten, maar ook bij oudere patiënten die gedurende 24 maanden werden behandeld. Op basis van deze gegevens hebben zij in 2015 een versnelde goedkeuring aangevraagd bij de FDA en een handelsvergunning aangevraagd bij de EMA. Daarnaast waren ze gestart met gefaseerde herdosering van patiënten in open-label extensiestudies met drisapersen (welke gestopt werden nadat de resultaten van de fase 3 studie bekend werden). De FDA meldde op 14 januari 2016 dat drisapersen momenteel niet klaar is voor goedkeuring.
Op 31 mei 2016 kondigde BioMarin aan dat ze hun aanvraag bij de EMA introkken. Ze hebben de ontwikkeling van drisapersen en ook andere AONs gericht op exon 44, 45 en 53, die in klinische ontwikkeling waren, stopgezet. Ze werken momenteel aan de ontwikkeling van effectievere en veiligere middelen voor exon skipping.
Stop codon readthrough medicatie
Ataluren en gentamicine
Huidige stand van zaken
Ataluren heeft conditionele goedkeuring ontvangen van de EMA for de behandeling van ambulante Duchenne patienten vanaf 4 jaar oud met een nonsense mutatie. Dit label is later uitgebreid naar 2 jaar en ouder. In 2023 heeft de EMA aangegeven de goedkeuring niet te verlengen. De Europeese commissie heeft deze beslissing in 2025 bekrachtigd. Ataluren zal waarschijnlijk van de markt gehaald worden in EMA landen. Deze keuze heeft geen impact op landen buiten de EU waar ataluren op de markt is.
Toepasbaarheid
Deze stop codon readthrough medicijnen werken alleen voor patiënten met een “stopsignaal”-mutatie (nonsense mutatie). Deze mutaties hebben geen invloed op de genetische code, maar introduceren een stopsignaal in het midden van het gen naast het stopsignaal aan het einde van het gen dat aangeeft dat de vertaling naar eiwit voltooid is. Dit is bij ~10-15% van de Duchenne-patiënten het geval. De geneesmiddelen kunnen ook gunstig zijn voor personen met stopcodons in andere genen.
Doel
Om de cel te dwingen het gemuteerde stopcodon te negeren en een compleet dystrofine-eiwit te produceren.
Achtergrond
Alle genen hebben een startsignaal en een stopsignaal, zodat de machine die genen omzet in eiwitten, weet waar te beginnen met de translatie en waar te eindigen. Soms kan een kleine mutatie een stopsignaal in het gen introduceren (naast het normale stopsignaal dat aan het einde van het gen aanwezig is). Dit type mutatie wordt een ‘nonsense mutatie’ genoemd. Normale stopsignalen verschillen over het algemeen enigszins van deze gemuteerde stopsignalen. Vergelijk het met een stopsignaal bij een druk kruispunt (normaal stopsignaal) en één op een snelweg (gemuteerd stopsignaal). Desondanks zal de cel het verkeerde stopsignaal volgen en de vertaling naar eiwit voortijdig stoppen. Er zijn medicijnen die het gebruik van nonsense mutaties onderdrukken, terwijl ze geen invloed hebben op de normale stopcodons. Het eerste medicijn dat werd geïdentificeerd om dit te doen in gekweekte cellen en Duchenne-muismodellen was gentamicine (een antibioticum uit de animoglycosiden klasse).
1. Klinische studie – gentamicine
Gentamicine is getest bij patiënten met Duchenne (studie 1 en 2), maar heeft nooit een overtuigend herstel van dystrofine laten zien.
Uitdaging 1
Naast het lage rendement is gentamicine toxisch bij langdurig gebruik (het kan de oren en de nieren beschadigen).
Oplossing 1
Screening van een groot aantal geneesmiddelen resulteerde in de identificatie van een medicijn zonder de toxische bijwerkingen dat ook in staat was om cellen te dwingen gemuteerde stopcodons te negeren. Dit medicijn wordt PTC124, ataluren of Franslarna™ genoemd en is ontwikkeld door PTC Therapeutics (VS). Het kan oraal worden ingenomen en resulteerde in herstel van dystrofine-expressie in gekweekte cellen en mdx-muizen.
Ataluren was voorwaardelijk goedgekeurd in de EU en is nog steed goedgekeurd in Brazilië, de UK en Rusland voor ammbulante Duchenne patienten met een nonsense mutate van 2 jaar en ouder.
2. Klinische studie – ataluren
Ataluren was veilig bij gezonde vrijwilligers. Een eerste studie met Duchenne-patiënten waarbij patiënten gedurende 4 weken werden behandeld met ataluren in verschillende dagelijkse doses toonde aan dat de behandeling goed werd verdragen en liet een verhoging van de dystrofine-expressie zien in behandelde patiënten. Om te testen of dit ook resulteerde in functionele verbetering op de lange termijn zijn klinische studies uitgevoerd in meerdere centra in de VS en Europa (studie 1 en 2).
Helaas leidde de behandeling niet overtuigend tot een functionele verbetering in een 6 minuten looptest in vergelijking met placebo-behandelde patiënten en daarom werden de studies gepauzeerd. Patiënten die betrokken zijn bij deze onderzoeken in de VS en Europa, kunnen zich inschrijven voor een open-label studie.
Na een gedetailleerde analyse van de gegevens en een verdere optimalisatie van de dosering, is ter bevestiging een nieuwe fase 3 studie bij 220 Duchenne-patiënten afgerond in Noord- en Zuid-Amerika, Azië, Australië en Europa. De met ataluren behandelde patiënten liepen gemiddeld 15 meter meer in zes minuten in vergelijking met de met placebo behandelde patiënten. In de vooraf gespecificeerde subgroep van patiënten (die tussen de 300 en 400 meter in zes minuten konden lopen aan het begin van de studie) liepen de met ataluren behandelde patiënten 47 meter verder dan de met placebo behandelde patiënten. Met ataluren behandelde patiënten presteerden ook beter in andere functionele tests. Zoals in eerdere studies ook was gezien, werd ataluren goed verdragen. Op basis van deze gegevens verlengde de EMA de voorwaardelijke goedkeuring en vraagde een nieuw bevestigend onderzoek aan. In dit onderzoek werden patiënten gedurende 72 weken behandeld met ataluren of placebo. De behandelde patiënten liepen bij de 6 minuten looptest 14 meter meer dan de placebogroep (significant verschillend). Tijdens de studie werden 12 met ataluren behandelde patiënten niet-ambulant versus 20 met placebo behandelde patiënten. Behandelde patiënten presteerden beter in de NSAA en getimede functietests.
Bovendien startte PTC Therapeutics een klinische studie om de dystrofineniveaus voor en na de behandeling te beoordelen. De resultaten zijn beschikbaar en suggereren suggereren een verhoogde dystrofine-expressie na de behandeling.
Sinds ataluren voorwaardelijk werd goedgekeurd in Europa, verzamelde PTC bewijs uit de praktijk van patiënten die (niet in het kader van klinische studies) behandeld worden met ataluren. Tot nu toe suggereert dit dat, in vergelijking met het natuurlijke ziektebeloop, patiënten behandeld met ataluren op latere leeftijd in een rolstoel terecht komen en ademhalingsproblemen krijgen.
Op basis van deze resultaten heeft PTC een aanvraag ingediend om volledige (in plaats van voorwaardelijke) markttoelating bij de EMA te verkrijgen en een handelsvergunning bij de FDA. Op 15 september 2023 kondigde de EMA aan dat het CHMP had besloten geen volledige markttoelating te verlenen en ook de voorwaardelijke goedkeuring voor ataluren niet te verlengen. PTC ging in beroep tegen dit besluit, waarna de EMA de gegevens opnieuw heeft geëvalueerd. Op 26 januari 2024 kondigde de EMA aan dat het CHMP, ook na herbeoordeling, aanbeveelt de voorwaardelijke goedkeuring niet te verlengen. De Europese Commissie heeft deze beslissing bekrachtigd.
Tegelijkertijd heeft PTC een nieuwe geneesmiddelenaanvraag ingediend bij de FDA, die momenteel wordt beoordeeld.
Lees verder